July 14, 2021
Here is the article. I like to spend 20 minutes to read carefully and learn more about clinical trial III stage and evaluate my position and risk.
Summary
- CEL-SCI announced successful Phase III clinical trial results of its drug Multikine, which can treat 155,000 newly diagnosed advanced primary head and neck cancer patients per year globally.
- Five-year survival: control arm 48.6%, Multikine arm 62.7% (N=380, p=0.0236, HR=0.68); in other words, 21,000 more patients would be alive after five years versus the current standard of care.
- Multikine had no safety issues, unlike any other cancer treatment.
- Multikine is the first treatment in 30 years for newly-diagnosed squamous cell head and neck tumors. As such, it serves an unmet medical need, which is important to FDA.
- FDA approval and multi-billion dollar commercial success is highly probable. The current market cap of ~$0.40 billion is unjustifiably low.
I. Introduction
CEL-SCI (NYSE: CVM) is a Virginia-based biotech company aimed at fighting squamous cell head and neck cancer. It just announced successful Phase III clinical trial results and it intends to seek FDA approval in the near future. CEL-SCI's Multikine is thus poised to be part of the standard of care (SOC) for 40% of newly-diagnosed advanced primary head and neck cancer patients, which is 155,000 patients per year globally.
We believe CEL-SCI shares should be in the high double-digits right now. Within 18 months, using conservative estimates of price and market penetration, we see a high probability of at least a $6B enterprise value ($113 per share fully diluted) following FDA approval. Below is a preview of a value calculation using some conservative assumptions:
TABLE 1:
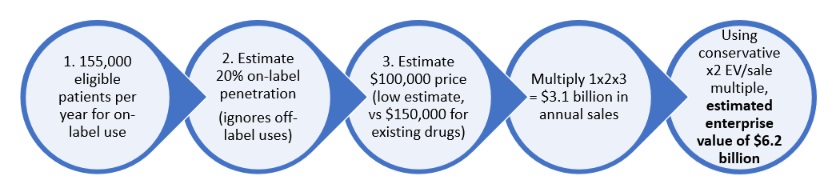
The current market value of $0.40 billion is far too low.
II. Multikine Is Designed To Address An Unmet Medical Need
All cancer is horrible, but advanced squamous cell head and neck cancer may be one of the most horrible. It is particularly deadly, with the SOC providing a 5-year survival of around 50%. The SOC generally requires surgery, i.e., the patient's tongue or parts of their face and neck may need to be cut out, and then either radiotherapy or chemoradiotherapy is usually given. As one oncologist stated:
Head and neck cancer is possibly the most horrific of all cancers. Not only does it take your life, but it takes your beauty, your voice and your dignity.
(Source: CEL-SCI Shareholder Letter dated July 7, 2021.)
In addition to its gruesomeness, treating head and neck cancer is especially difficult. (Source.)
There has not been a new first-line treatment for advanced squamous cell head and neck cancer in decades, meaning a treatment given right after diagnoses. Major drug manufacturers are keenly interested in this unmet medical need, and have recently tried and failed to develop drugs for it:
TABLE 2:
| Pfizer & Merck | Bavencio | Failed March 2020 |
| AstraZeneca | Durvalumab | Failed February 2021 |
| Boehringer | Afatinib | Failed June 2019 |
| Glaxo | Feladilimab | Failed April 2021 |
Two of the most successful cancer drugs in history – MRK's Keytruda and BMY's Opdivo – have been deployed to fight head and neck cancer, but not as a first-line defense. Rather, they are considered "late stage," i.e., indicated only for patients whose cancer has spread or returned and cannot be removed by surgery. In addition, Keytruda and Opdivo are toxic, and studies show that they extend median overall survival by just a few months. (See Keytruda clinical study results and Opdivo results here.) Keytruda and Opdivo sales still total many billions of dollars annually. (See Keytruda sales and Opdivo sales.)
CEL-SCI's Multikine is different. It is given as a first-line treatment for newly-diagnosed primary advanced head and neck cancer patients. Multikine would thus serve an unmet medical need that is outside the approved indications for Keytruda and Opdivo, shown in TABLE 3 below. Serving an unmet medical need is one of the key factors that FDA considers in determining whether to approve a drug.
TABLE 3:

(Sources: CEL-SCI Shareholder Letter dated July 7, 2021; Keytruda Indications for Use; Opdivo Indications for Use.)
III. CEL-SCI's Clinical Trial of Multikine
Study Design
CEL-SCI's pivotal clinical trial of Multikine on 928 patients was unusually large in order to accommodate the different treatments in the SOC control arm:
- Enrollment. All patients meeting eligibility criteria are enrolled.
- Pre-surgical treatment. In the weeks before surgery, the study randomized eligible patients into three groups. First, patients were given Multikine and a supplement called "CIZ" (which contains an immunosuppressant, an anti-inflammatory, and a vitamin). Second, patients were given Multikine alone. Third, patients were not given Multikine prior to surgery. The overall study was conducted only between the first and third groups.
- Surgery. Per the National Comprehensive Cancer Network Clinical Practice Guidelines in Oncology (NCCN Guidelines), all study participants would have surgery to remove the tumor.
- Post-surgical SOC. Following surgery, per NCCN Guidelines, the patient's condition was assessed by the physician to determine if they were at a lower or higher risk for tumor recurrence. Lower-risk patients received only radiotherapy. Higher-risk patients received chemoradiotherapy. Each patient would follow one of these two SOC treatment paths.
The graphic below depicts this flow of patient treatment:
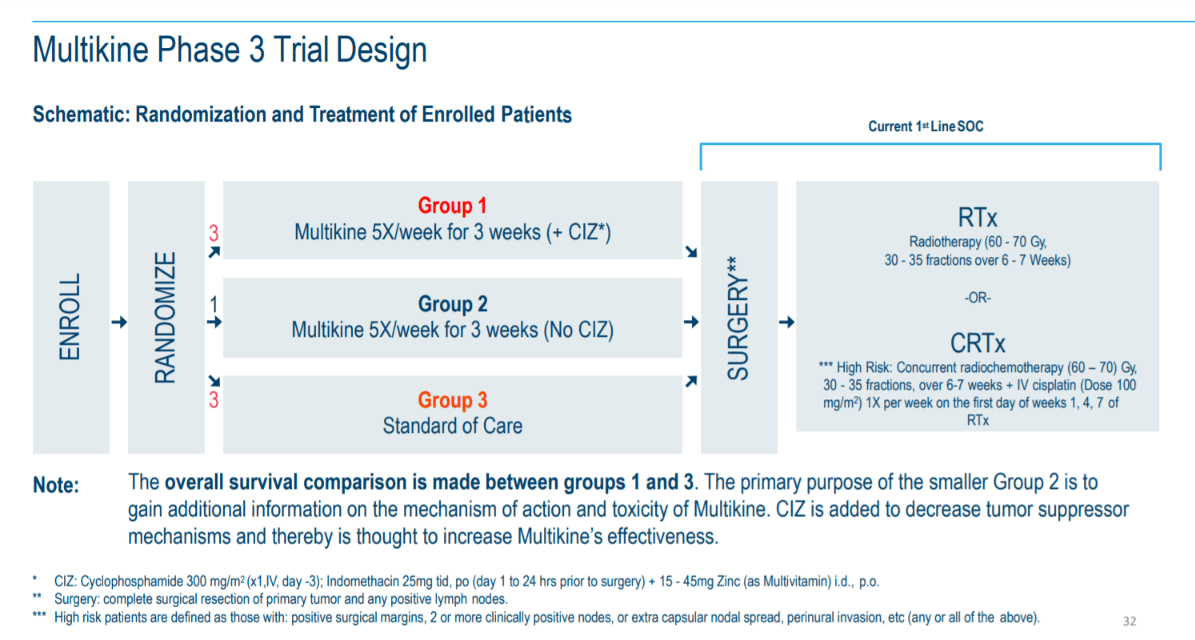 (Source: Cel-Sci Corporate Presentation dated June 10, 2020.)
(Source: Cel-Sci Corporate Presentation dated June 10, 2020.)
On the far right, you can see "RTx" for radiotherapy and "CRTx" for chemoradiotherapy. We will use these shorthands for the remainder of this article.
As CEL-SCI noted in its recent shareholder letter, the clinical protocol developed at the start of the study provided for analyses of Multikine's clinical efficacy in at least three separate ways: (i) in both the RTx and CRTx arms together; (ii) in the RTx arm alone; and (iii) in the CRTx arm alone.
In order to provide sufficient statistical power, the protocol required 298 deaths to occur out of the 928 enrolled patients, and the 298th death occurred in April 2020. The data then needed to be audited, collected, reviewed, and locked for analysis. This process, known as "data lock," was performed by two well-known clinical research organizations: Ergomed and ICON plc, an acclaimed biometrics company that recently won five best-in-class awards. Data lock required in-person visits to hospitals in order to inspect and verify medical records, and this was substantially delayed due to COVID. Before data lock was complete and before data analysis could begin, CEL-SCI and ICON signed off on a statistical analysis plan that would govern ICON's data analysis. In December 2020, CEL-SCI announced that data lock had been completed. All this time, CEL-SCI remained blinded to the data.
Study Results
The results of the study showed that Multikine would save thousands of lives. Specifically, in the 380-patient RTx arm, 62.7% of patients who received Multikine were still alive at year five, while only 48.6% survived that long with RTx alone. (Source: CEL-SCI Shareholder Letter dated July 7, 2021.)
Because the RTx patient population would include 155,000 head and neck cancer patients every year, Multikine's 14.1% overall survival benefit would mean that 21,000 patients could be saved every year. That is the entire crowd of an average pre-COVID major league baseball game alive vs dead.
The survival benefit showed a statistical power of p=0.0236, well below the study's criterion of p<0.05. (The lower the p-value, the stronger the significance of the data.) The hazard ratio of 0.68 was lower than the study's criterion of 0.72 (lower is better). The hazard ratio means that patients in the RTx arm who received Multikine survived longer than those who did not. The table below summarizes these results:
TABLE 4:
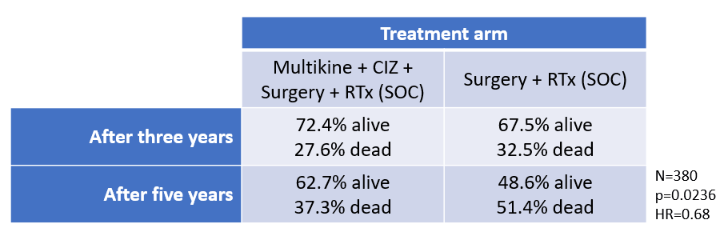 (Source: Shareholder Conference July 1, 2021.)
(Source: Shareholder Conference July 1, 2021.)
Not shown in TABLE 4 is the analyses of (i) the CRTx arm or (ii) the entire study population (all RTx and CRTx patients together). CEL-SCI reported that these analyses did not meet the primary endpoint of 10% overall survival benefit in those populations. But this does not mean that Multikine failed. Multikine was successful in the RTx arm, which represented about 40% of the study population, a substantial portion. Would it have been great to see strong efficacy in both treatment arms? Of course. But a significant positive result in a 380-patient treatment arm is still highly beneficial, particularly with a non-toxic drug. It is likely that the FDA will think so, too, since their mandate is to approve drugs that provide benefits to patients that outweigh the risks.
Multikine's "no safety issues" sits in sharp contrast to CRTx, which is highly toxic by its very nature. Many patients cannot tolerate CRTx, and we suspect that nearly everyone personally knows of someone who has suffered its ravages. For many who eventually die from cancer, it can be unclear whether it was the cancer or the chemo that ended their life. Multikine's efficacy without any such toxicities represents a landmark achievement in cancer treatment.
IV. FDA Approval Is Highly Likely
CEL-SCI announced that the clinical trial results showing safety and efficacy with strong statistical power will form the basis of a Biologics License Application (BLA) to be filed with FDA. We believe it is highly probable that the BLA will be approved, and we outline ten reasons for this belief below.
First, the FDA requires clinical trial analyses to be done prospectively and while the company is blinded. CEL-SCI has stated that it satisfies these criteria:
The analysis for the successful treatment arm was pre-specified in the study protocol and also the study Statistical Analysis Plan (SAP). These documents specified that we would analyze and present to the FDA not only the combined results of the two treatment arms but also each individual treatment arm . . . .
The surgery plus radiation arm of the study was pre-specified and is in fact one of the 2 potential treatment arms per the NCCN Treatment Guidelines for this disease. Analysis of any of the treatment arms on their own was always a part of the statistical analysis plan, and it was pre-specified before database lock and before anyone began analyzing the data. . . .
To summarize again, all analyses were pre-specified in the protocol and SAP and were performed only following database lock and prior to unblinding to the study data. Therefore, we are permitted to use the Multikine plus surgery and radiation data for FDA approval submission.
(Source: CEL-SCI Shareholder Letter dated July 7, 2021.)
Thus, there was no "data-mining" or "data dredging" after the fact, as some have alleged.
Second, ICON would not have conducted the RTx-arm analysis if they had not been pre-specified in the protocol and statistical analysis plan before data analysis began. To have done otherwise would have been to put ICON's award-winning reputation at risk (and potentially expose it to legal liability) by improperly data-mining. It makes no sense to suggest that ICON would commit commercial suicide for the sake of one clinical study. This corroborates CEL-SCI's statements that the analysis was indeed prospective, pre-specified in the protocol and the statistical analysis plan, and not data-mined.
Third, CEL-SCI explained that ICON's analysis was further double-checked by an independent statistician. With its own independent reputation and business on the line (indeed, its credibility at the FDA is the life-blood of its business), the statistician denied any data-mining and affirmed the reliability of the data for FDA. CEL-SCI quoted the statistician's entire statement in its July 7, 2021 shareholder letter. The independent statistician's explanation provides further corroboration for the reliability of CEL-SCI's data for FDA approval.
Fourth, Multikine's results are favorable when compared to the studies for Keytruda and Opdivo. Keytruda was approved in 2016 for head and neck cancer that has spread or come back after chemotherapy. This was the first immunotherapy approval for head and neck cancer, and it was based on a subgroup analysis of only 174 patients, much smaller than CEL-SCI's N=380. In the KEYNOTE-012 study (page 73), only 16% responded to the drug, and yet this was still a tremendous achievement, prompting the FDA to grant accelerated approval for the new indication.
Opdivo's study population of N=361 was also similar to CEL-SCI's N=380. Opdivo's survival benefit was a median life extension of only about 2.4 months. By contrast, with Multikine, 14.1% more patients were alive than dead at year five. Opdivo also had a higher (worse) hazard ratio than CEL-SCI.
Critically, both Keytruda and Opdivo have horrible toxicity. For instance:
In KEYNOTE-012, KEYTRUDA was discontinued due to adverse reactions in 17% of 192 patients with HNSCC. Serious adverse reactions occurred in 45% of patients.
(Source.)
Multikine has no such toxicity.
Thus, given the regulatory success of Keytruda/Opdivo as comparators, it is likely that FDA will look favorably on Multikine.
TABLE 5:
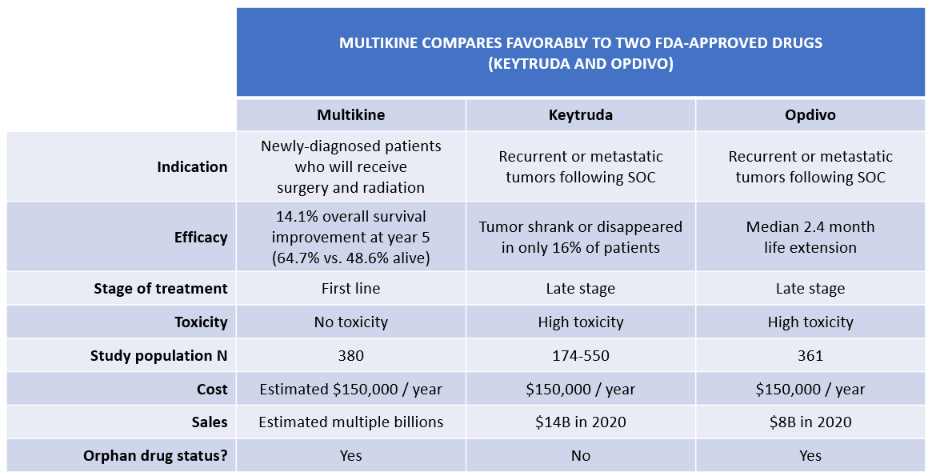
Fifth, Multikine's FDA approval is also supported by the fact that it satisfies an unmet patient need, as there has been no new first-line treatment for advanced head and neck cancer in over 30 years – until now. Serving an unmet medical need is important to the FDA, as shown in recent guidance.
Sixth, CEL-SCI stated it would seek approval only for patients indicated for surgery and radiation. It is irrelevant, therefore, whether Multikine did not work in a different group of patients (i.e., those who also received chemotherapy). What matters to the FDA is whether CEL-SCI can prove safety and efficacy in the "intended population."
Seventh, CEL-SCI stated that it could determine reliably who would receive RTx and who would require CRTx, such that physicians would know in advance which patients should receive Multikine:
We have determined that it is possible to select the population that would receive the Multikine benefit at their time of diagnosis. We have vetted this with a number of expert physicians in the field and they agreed with the feasibility of the proposed pre-selection methodology.
(Source: CEL-SCI Shareholder Letter dated July 7, 2021.)
Eighth, the FDA currently has a policy in favor of approving safe drugs, particularly when there is an unmet medical need, as with the recent Biogen (BIIB) Alzheimer's drug Aduhelm. A recent analyst report agrees that this factor weighs in CEL-SCI's favor. In addition, as we all know, President Biden's son died of brain cancer, and the President has spoken out about the need to improve treatments. The Administration's outspoken policy is likely to be a thumb on the scales in favor of approval.
Ninth, Multikine appears to satisfy FDA guidance for approval:
FDA approval of a drug means that data on the drug’s effects have been reviewed by CDER, and the drug is determined to provide benefits that outweigh its known and potential risks for the intended population.
(Source.)
Multikine is effective with no safety issues in the intended population, i.e., patients who will receive surgery and RTx.
The FDA guidance points to three important factors, all of which support the likelihood of Multikine's approval:
FDA reviewers analyze the condition or illness for which the drug is intended and evaluate the current treatment landscape, which provide the context for weighing the drug’s risks and benefits. For example, a drug intended to treat patients with a life-threatening disease for which no other therapy exists may be considered to have benefits that outweigh the risks even if those risks would be considered unacceptable for a condition that is not life threatening."
Multikine is intended to treat patients with life-threatening disease (cancer), and there is currently no other first-line therapy for newly-diagnosed advanced squamous cell head and neck tumors besides surgery. Multikine also has zero safety issues. Thus, the standard for approval will be easier for Multikine than for drugs that do not treat life-threatening illnesses or for drugs that are toxic.
"FDA reviewers evaluate clinical benefit and risk information submitted by the drug maker, taking into account any uncertainties that may result from imperfect or incomplete data. Generally, the agency expects that the drug maker will submit results from two well-designed clinical trials, to be sure that the findings from the first trial are not the result of chance or bias. In certain cases, especially if the disease is rare and multiple trials may not be feasible, convincing evidence from one clinical trial may be enough. Evidence that the drug will benefit the target population should outweigh any risks and uncertainties."
According to this standard, CEL-SCI will be permitted to rely on a single pivotal clinical trial for Multikine, because it treats a "rare" disease, which is one that affects fewer than 200,000 people in the United States. Squamous cell cancer of the head and neck affects 60,000 people annually in the United States. Moreover, Multikine has already been accorded Orphan Drug status, which is given to drugs "intended for the treatment, prevention or diagnosis of a rare disease...." Given Multikine's 14.1% overall survival benefit for the intended population with no safety issues, there is "evidence that the drug will benefit the target population" that "outweigh[s] any risks and uncertainties."
"All drugs have risks. Risk management strategies include an FDA-approved drug label, which clearly describes the drug’s benefits and risks, and how the risks can be detected and managed. Sometimes, more effort is needed to manage risks. In these cases, a drug maker may need to implement a Risk Management and Mitigation Strategy (REMS)."
Although the FDA always weighs benefits and risks, when it comes to cancer, the FDA does not require much efficacy even in the face of serious adverse events. Consider Opdivo, which for head and neck cancer provides a median life extension of just 2.4 months and yet is highly toxic. With Keytruda, only 16% of patients even responded to treatment and there were also serious adverse events. Yet the FDA approved these drugs. As discussed, when seen against this backdrop, Multikine's approval seems highly probable.
Special approval pathways also appear available to Multikine, including Fast Track, Breakthrough Therapy, Accelerated Approval, and Priority Review:
TABLE 6:
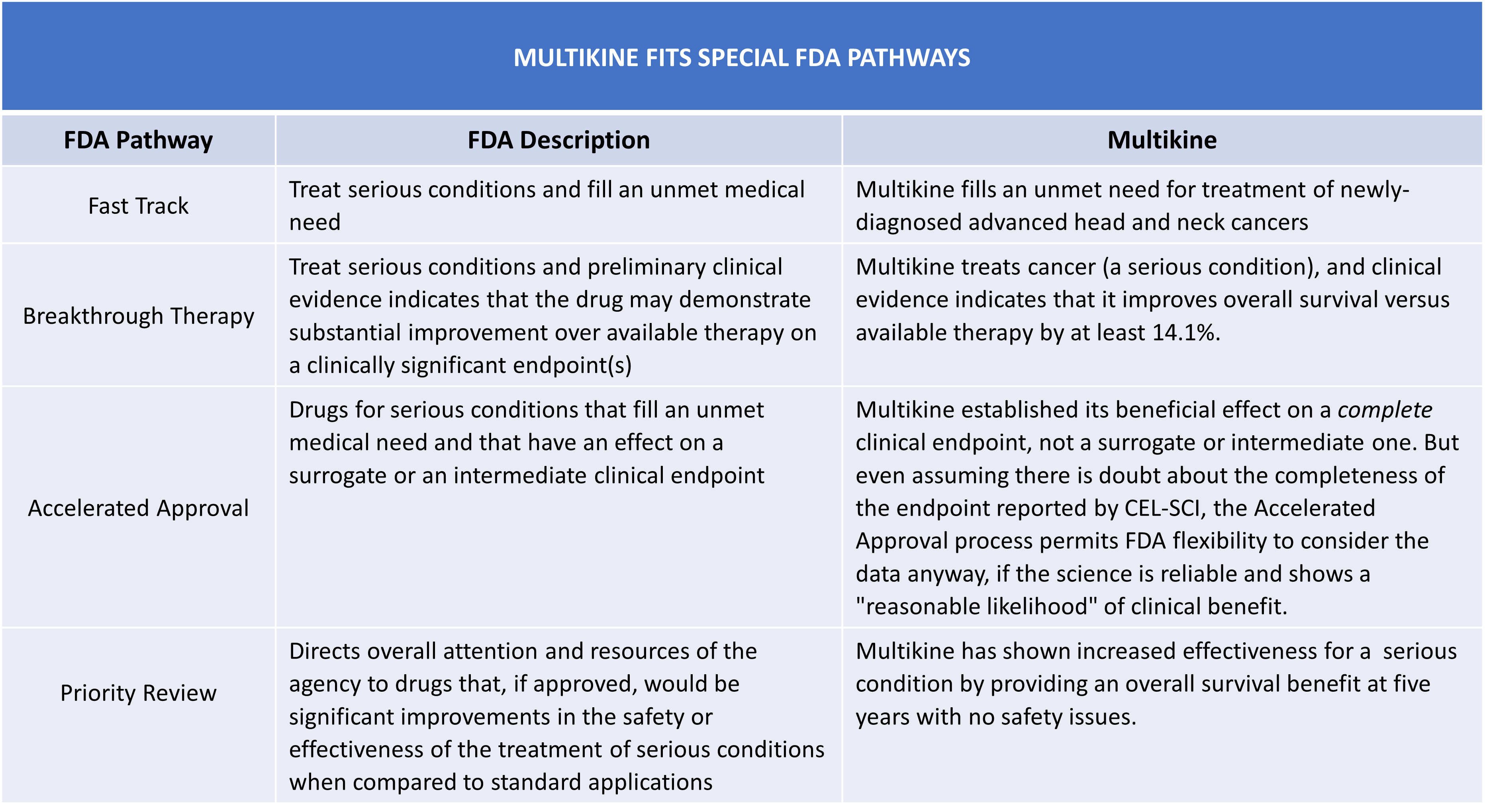 Accordingly, FDA guidance supports the likelihood of Multikine's approval.
Accordingly, FDA guidance supports the likelihood of Multikine's approval.
Tenth, because it would have been unethical to deny any patient the SOC, CEL-SCI could not have designed a different study. All of the patients had to receive the SOC, which for some included CRTx. If failure of the primary endpoint involving the combined RTx + CRTx populations meant that one could not also analyze the overall survival benefit in the RTx and CRTx populations separately, then there could never be an approval for a drug that helped only one of those populations (here, RTx). This is illogical and inconsistent with FDA's mandate to approve drugs that "provide benefits that outweigh its known and potential risks for the intended population." Given the success Multikine showed in the RTx group with no safety issues, it seems highly probable that FDA will want to get this drug to these patients.
In view of at least these ten reasons, we believe FDA approval for the RTx indication is highly probable. (Additional reasons supporting the likelihood of FDA approval can be found in this article and this article as well.)
V. CEL-SCI Would Have A Multi-Billion Dollar Value If Multikine Were Approved
Assuming Multikine is approved for patients indicated for RTx after surgery, we can estimate the potential value of CEL-SCI post-approval. The population indicated for RTx after surgery is 155,000 patients per year globally. The price of Keytruda and Opdivo is about $150,000 per year. Using these figures as benchmarks, we built a table of estimated annual revenues based on % penetration of on-label use (i.e., % of 155,000 patients) and the price of Multikine (we think it will sell for the same $150,000 as Keytruda and Opdivo, but to be conservative, we will look at lower prices):
TABLE 7:
 (Source: author's analysis, where each cell is 155,000 multiplied by percent penetration multiplied by price per year.)
(Source: author's analysis, where each cell is 155,000 multiplied by percent penetration multiplied by price per year.)
The blue region represents penetration from 15% to 35% of patients within the RTx indication and prices from $70,000 to $130,000. As you can see, if approved, Multikine would be a multi-billion dollar product.
Below, we applied a very low EV-to-sales multiple of just 2x to estimate the enterprise value based on this revenue stream:
TABLE 8:
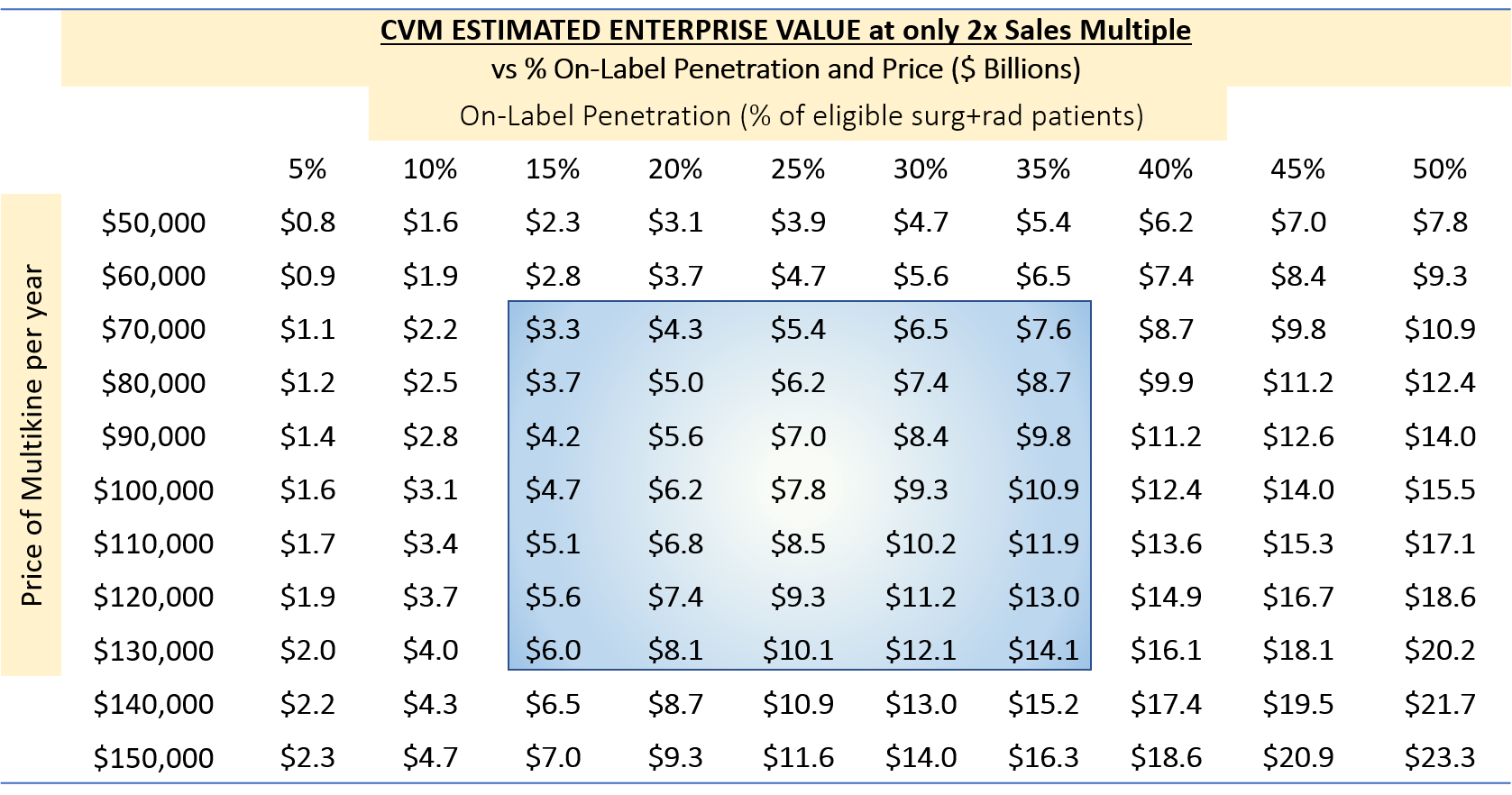 (Source: author's analysis, where each cell is from TABLE 6 multiplied by 2.)
(Source: author's analysis, where each cell is from TABLE 6 multiplied by 2.)
The enterprise value for these scenarios would be $3.3 billion to $14.1 billion. We note that the 2x multiple is extremely low for a successful biotech buy-out. A 4x multiple would be more realistic, which would double the figures above.
Finally, we can show these enterprise values in terms of share price:
TABLE 9: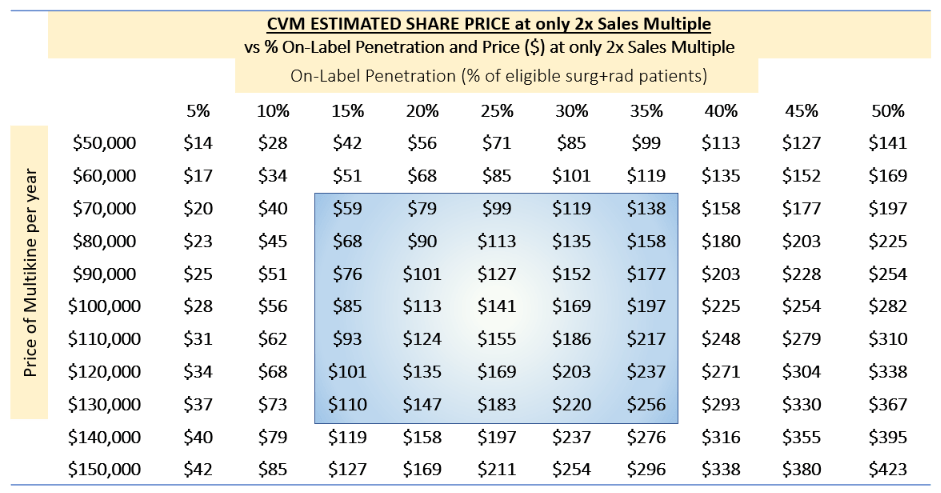
(Source: author's analysis, where each cell is TABLE 7 multiplied by 1000 divided by 55, reflecting $1B/55M shares outstanding, fully diluted.)
Under the same range of assumptions, CEL-SCI's share price range from about $59 to about $256 if Multikine were approved.
We believe the above figures are conservative for the following five reasons:
- There is no reason to suspect that Multikine would cost less than Keytruda or Opdivo at $150,000 per year. At that price, the mid-case value would exceed $11B.
- We do not account for off-label use, which we expect to be substantial, given Multikine's safety profile.
- Average enterprise value / sales multiples for successful biotechs are generally much higher than 2x.
- Multikine might obtain FDA approval for additional indications in the future (e.g., to treat other cancers), just as Keytruda and Opdivo have done.
- In CEL-SCI's Phase II trial of Multikine, "10.5% of patients had no remaining cancer cells (by pathology) following 3 weeks of Multikine alone." If this occurred in the Phase III trial, even on a smaller scale, it would still be monumental; imaging curing people of cancer within three weeks of a few non-toxic injections. We know that 76 patients in the Phase III study did not receive either RTx or CRTx, and it is unclear why such a large number of patients received no further treatment after surgery, especially where RTx has few contraindications. It is possible that, as with the Phase II trial, the tumors of some patients had very positive responses to Multikine prior to surgery.
VI. Conclusion: CEL-SCI Is Significantly Undervalued Now
Because the company has more than $47 million in cash, the only real barrier to commercialization is FDA approval. Indeed, insiders have recently bought $250,000 worth of CEL-SCI stock, including $200,000 from its CEO, Geert Kersten. This reflects the insiders' confidence in obtaining FDA approval and in the ultimate success of the company.
But we don't need to rely on the insiders. We have considered all the evidence presented in this article and conclude for ourselves that FDA approval is highly likely. We can calculate that the value of CEL-SCI post-approval should be multiple billions of dollars. The current value of $0.40B is thus far too low.
Comparing our estimates of CEL-SCI's post-FDA approval value to the current stock price, we can infer that the market is predicting a roughly 10% to 15% chance of FDA approval. We believe this is well below any rational assessment, and, therefore, we view the current price as irrationally low.
There are still risks for the company. The FDA might not approve the drug without additional clinical trials, though this appears unlikely given the trial results, no safety issues, and the prior precedents of Keytruda/Opdivo. Even if the drug is approved, there is a risk that another drug would supplant Multikine as a first line treatment, though we are unaware of any pending clinical trials in that regard. There is also a risk that the company needs cash before FDA approval, though this is also unlikely given how the cash they currently have on hand. Finally, there is a risk that CEL-SCI cannot provide adequate manufacturing capacity, though is more of a limitation on upside.
We believe the likelihood of approval is at least 75% in view of the reasons discussed above. With a 75% risk-adjustment on a $60 per share post-approval valuation (a low-case scenario), the current share price should be around $45 right now. If you believe that the post-approval value of CEL-SCI would exceed $7.5B (a mid-case scenario, using the low 2x multiple), then the share price should be more than $100 right now. If you are still skeptical, you can even cut these figures in half (e.g., assume a 35% probability of FDA approval) and arrive at a risk-adjusted present value that is still many times higher than the current $9 share price.
As the numbers are analyzed and more people come to understand the positive trial results, we expect the share price will rise substantially in the near term. CEL-SCI presents a tremendous opportunity for significant upside in the next 18 months. We are long.
No comments:
Post a Comment